A home finger-prick test promises to catch hidden cardiometabolic risks in young adults, if paired with hands-on guidance and smarter support systems.
Study: Feasibility of self-administered dried blood spot collection for…

A home finger-prick test promises to catch hidden cardiometabolic risks in young adults, if paired with hands-on guidance and smarter support systems.
Study: Feasibility of self-administered dried blood spot collection for…

Microsoft will end support for Windows 10 on 14 October 2025, stopping security updates, technical support, and new features for the operating system.
Following the end-of-life (EoL) date, Windows 10 users will need to consider their…
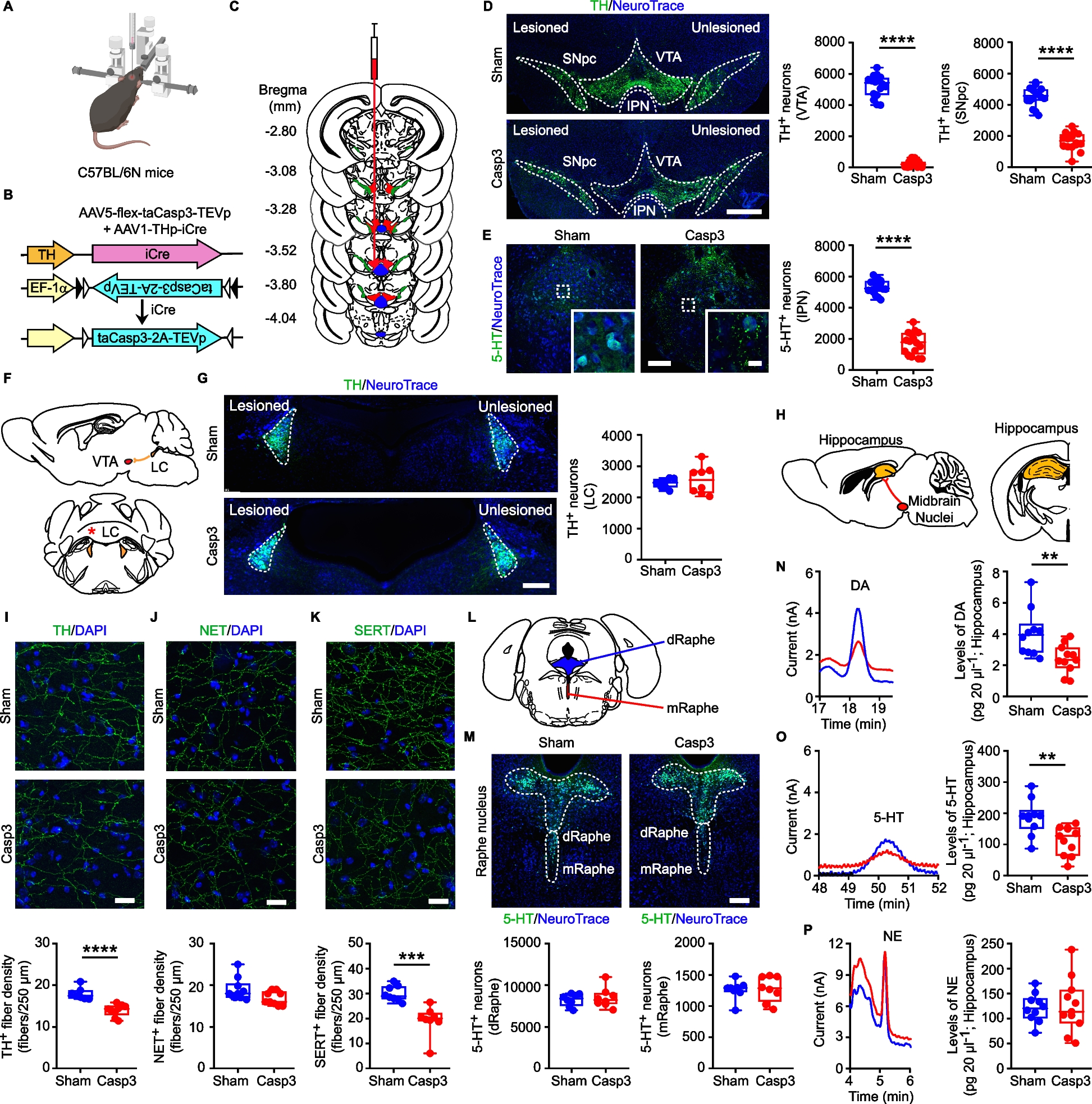
Experiments complied with the ARRIVE procedures and the ethical guidelines of the European Council Directive (2010/63/EU). Experimental approval was obtained from the Italian Ministry of Health. Male and female C57BL/6N (Charles River, Italy) and female B6.129P2(Cg)-Cx3cr1tm1Litt/J (CX3CR-1GFP knock-in/knock-out; Jackson Laboratory Strain #:005582) mice were used at 2–3 months of age. Heterozygous male Tg2576 mice (Taconic #APPSWE—Model 1349 tg/wt; [41]) and their Wild-Type (WT) littermates (mice with the same strain and genetic background of Tg2576, negative for APPSWE overexpression—Taconic #APPSWE—Model 1349 wt/wt) were used at 6–7 months of age.
All mice were housed with ad libitum food and water, with a 12 h light/dark cycle. Mice were housed in cages of 3–4 individuals and all cages were equipped with the same environmental enrichment.
Mice were anaesthetized with Rompun (20 mg/mL, 0.5 mL/kg; Bayer) and Zoletil (100 mg/mL, 0.5 mL/kg; Virbac; intraperitoneally i.p.) and positioned in a stereotaxic apparatus.
For Caspase-3 (Casp3) mice (C57BL/6N, CX3CR-1GFP or Tg2576 mice), we infused a mix of Adeno-Associated Viruses (AAVs; 0.5 µL each, flux 80 nL/min [42]: (i) AAV1-THp-iCre (5 × 1012 viral particles/mL; Vector Biolabs) and (ii) AAV5-flex-taCasp3-TEVp (4.2 × 1012 viral particles/mL; UNC Vector core, gift from Nirao Shah) in the left VTA (AP: −3.2, ML: −0.35, DV: −4.4; 43), to lesion Tyrosine Hydroxylase-positive (TH+) neurons in the VTA and Substantia Nigra pars compacta (SNpc) and 5-HT+ neurons in the interpeduncular nucleus (IPN). Sham mice were injected only with the AAV1-THp-iCre virus.
In a separate cohort of C57BL/6N mice, the AAV1-THp-iCre + AAV5-flex-taCasp3-TEVp mix (0.125 µL each, flux 25 nL/min) were injected in the IPN (thereafter: Casp3IPN; AP: −3.5, ML: 0, DV: −4.7; [43]) to lesion the 5-HT+ neurons of the IPN while leaving intact the TH+ neurons of the VTA/SNpc. Sham mice were injected only with the AAV1-THp-iCre virus (ShamIPN).
To confirm the ectopic expression of TH promoter-driven AAVs, a set of C57BL/6N mice were infused in the VTA with a mix (0.5 µL each, flux 80 nL/min) of (i) AAV1-THp-iCre and (ii) AAV5-EF1α-DIO-eYFP (4.0 × 1012 viral particles/mL; UNC Vector core, gift from Karl Deisseroth). eYFP expression was then analyzed with confocal microscopy (see below).
For 6-hydroxy-dopamine (6OHDA) lesion (C57BL/6N mice), 6OHDA (Sigma-Aldrich; 7.6 mg/mL, calculated as free-base) was dissolved in 0.2 mg/mL ascorbic-acid (Tocris) prepared in 0.9% saline, and continuously kept on ice. Each mouse was injected with 2.5 μg in 0.4 µL (flux 40 nL/min) unilaterally in the left VTA. Thirty minutes before surgery, mice received 10 mL/kg (2.85 mg/mL as free base, i.p.) of the norepinephrine (NE) reuptake-inhibitor desipramine-hydrochloride (Sigma-Aldrich), to prevent NE fiber degeneration. Control mice were injected with Saline.
C57BL/6N and CX3CR-1GFP mice received the intracerebral injection at 2 months of age. Tg2576 mice were injected at 6 months of age. All mice were used 30 days following injection to ensure steady-state lesion. A separate set of Casp3 and Sham were used at 72 h following lesion; in these mice we examined the levels of Casp3 protein in the midbrain (see below).
For retrograde labelling, red retrobeads (120 nL; 12 nL/min; Lumafluor) were injected bilaterally in the Nucleus Accumbens (NAc) medial shell (AP: 1.8, ML: ± 0.4, DV: 4.1), NAc core (AP: 1.5, ML: ± 0.9, DV: 4.1) and dorsal striatum (AP: 1.0, ML: ± 1.6, DV: 2.3). Mice were examined 1 month after surgery.
For all infusions we used 1 μL Hamilton syringes (Model Neuros7001) mounted on a Pump-11 Elite Nanomite (Harvard Apparatus). When possible, the accuracy of the injection site was controlled by immunofluorescence analysis; mice with misplaced injection were excluded.
Animals were injected with L-DOPA (i.p., 10 mg/kg; Sigma-Aldrich) plus benserazide (12 mg/kg; Sigma-Aldrich) [44], or with A68930-hydrochloride (5 mg/kg; Santa Cruz), once a day for 4 or 7 days, respectively; 0.9% saline was used as control (vehicle); experiments were performed 1 h after the last injection.
Fluoxetine-hydrochloride (30 mg/kg; Tocris) was dissolved in water and delivered ad libitum in drinking bottles wrapped in tin-foil for 30 days, starting immediately after surgery. Normal drinking water was used as control.
Anaesthetized mice (Rompun/Zoletil) were transcardially-perfused with Phosphate Buffer (PB; 0.1 M, pH 7.4) followed by 4% paraformaldehyde in PB. Brains were postfixed in 4% paraformaldehyde for at least 4 h, dehydrated and cryoprotected in 30% sucrose in PB at 4 °C until sinking. Thirty μm-thick coronal sections were cut with a cryostat, and slices were collected in PB-sodium azide 0.02%. All analyses were performed in the left hemisphere, ipsilateral to the lesion.
Slices were incubated with primary antibodies in PB containing 0.3% Triton X-100 overnight at 4 °C for TH, Iba1, GFAP, hippocampal 5-HT transporter (SERT) and striatal TH/DA transporter (DAT)/SERT staining, or 3 nights for IL-1β/Iba1, IL-18/Iba1, IL-18 Receptor (IL-18R)/GFAP, p-Nuclear Factor kappa B (p-NFκB)/S100β and Microtubule-Associated Protein 2 (MAP2).
For 5-HT, slices were incubated with primary antibodies in PB containing 0.5% Triton X-100 and 10% donkey-serum for 2 nights at 4 °C.
For hippocampal TH+/NE transporter-positive (NET+) fibers, sections were incubated in citrate buffer (10 mM sodium-citrate, pH 6.0 containing 0.05% Tween-20; 20 min, 75 °C), rinsed in PB, immersed in blocking solution (5% donkey serum, 0.2% Triton X-100 in PB; 1 h, RT), and incubated with primary antibody in the same solution (overnight at 4 °C; [45]).
For NLRP3/Iba1/GFAP and NLRP3/Iba1 staining, sections were pretreated with 50% methanol (15 min, RT) immersed in blocking solution (3% bovine serum albumin, 0.1% Triton X-100 in PB, 30 min, RT) and incubated with primary antibodies in PB with 0.1% Triton X-100 (2 nights, 4 °C).
For Iba1/CD68, slices were permeabilized using 0.5% Triton X-100 in PB (45 min, RT), incubated in blocking solution (2% bovine-serum albumin, 0.5% Triton X-100 in PB; 1 h, RT) and exposed to primary antibodies in blocking solution (2 nights, 4 °C).
For C3/GFAP, slices were immersed in blocking solution (5% donkey serum, 0.1% Triton X-100 in PB; 1 h, RT) and then incubated with primary antibodies in 1% donkey serum, 0.1% Triton X-100 in PB (overnight, 4 °C).
For C3aR/Iba1/NeuroTrace analysis, slices were incubated in blocking solution (3% bovine-serum albumin, 5% goat serum, 0.5% Triton X-100 in PB; 1 h, RT) and then with primary antibodies in blocking solution (overnight, 4 °C).
For tau staining (AT8), slices were pretreated with 50% methanol (15 min, RT), immersed in blocking solution (3% bovine serum albumin, 0.1% Triton X-100 in PB) with M.O.M.® (Mouse on Mouse) Blocking Reagent (1:1000; Vector laboratories, #MKB-2213–1; 1 h, RT) and then incubated with primary antibodies in 0.1% Triton X-100 in PB (2 nights, 4 °C).
For Aβ staining (6E10), sections were pretreated with M.O.M.® (1:1000; 2 h, RT) diluted in permeabilization solution (PB with 0.3% Triton X-100) and incubated with primary antibodies overnight at 4 °C in permeabilization solution.
For eYFP expression, coronal sections containing the midbrain, Raphe and LC were stained with TH/5-HT, 5-HT and TH, respectively as described above.
For every immunofluorescence protocol, after primary antibody, slices were incubated with secondary antibodies in the same solution of primary antibody (2 h, RT) and counterstained with Blue-Fluorescent Nissl-Stain (NeuroTrace 1:200; Invitrogen) or DAPI (1:1000, Serva).
After mounted, slices were examined using a Nikon Eclipse Ti2 confocal microscope. The labeling specificity was confirmed by omission of primary antibodies and use of normal serum instead (negative controls). For quantitative analysis, images were processed simultaneously and analyzed with Fiji-ImageJ (http://imagej.nih.gov/ij/): after 8-bit conversion and background subtraction, the signal was quantified by measuring the relative fluorescence intensity. The F/A ratio defines mean fluorescence intensity (F) over surface area (A).
For analysis of fiber density and intensity, and protein levels, images were acquired with a 20x-objective by Z-stacks, then processed by maximum-intensity projection. All samples were captured with identical Z-stack thickness and laser settings within each analysis.
TH+/DAT+/SERT+ fiber intensity was quantified by setting 16 frames (100 × 100 pixel). The total fiber number per 250 µm was counted manually [45, 46].
CD68, NLRP3, IL-1β and IL-18 protein levels were measured within Iba1+ cells.
Automated total cell counts and analysis of overlapping regions between markers to assess colocalization were performed in Imaris XT software (Bitplane AG, Oxford Instruments, Abingdon-on-Thames, UK). For all double-positive cell counting analyses, the Surfaces function was used. Briefly, after applying background subtraction and automatic thresholding, to retain only objects within lower and upper threshold Limits, and splitting of touching objects to separate individual cells, colocalization between markers was quantified using the Overlapped Volume Surfaces option. Complement component 3 (C3) levels were quantified within GFAP+ cells, while MAP2 intensity was quantified by setting 12 frames (90 × 90 pixel) over the stratum radiatum. Intracellular Aβ levels were quantified by setting 10 randomly-distributed frames on the hippocampal CA1 pyramidal layer (70 × 70 pixel).
To analyze the number of Aβ plaques and the AT8+ area in the hippocampus, we acquired Z-stack large images with a 20x-objective. The mean number of 6E10+ plaques in the hippocampus from at least 3 slices/animal was quantified. To calculate AT8+ area, the hippocampus was manually defined, and automatic brightness thresholding was used to delineate the positive area. Hippocampal AT8+ area was expressed as ratio of total area analyzed in each slice/animal (modified from [47]). Intracellular AT8 levels were quantified by Z-stack with 60x-oil objective and by setting 1 frame (20 × 20 pixel).
For all analyses, quantification was done on 4 slices/mouse. Data were then averaged per mouse for figures and statistical analysis.
Primary antibodies: 5-HT (1:500; ImmunoStar #20080; RRID:AB_572263), AT8 Ser202/Thr205 (1:200; Invitrogen #1020; RRID:AB_223647), C3 (1:300; Novus Biologicals #NB200-540; RRID:AB_2744548), C3aR (1:200; HycultBiotech #HM3028; RRID:AB_2131309), CD68 (1:400; Biorad #MCA1957; RRID:AB_322219), DAT (1:400; Chemicon #MAB369; RRID:AB_2190413), GFAP (1:1000; Millipore #AB5804; RRID:AB_2109645), GFAP (1:1000; DAKO #Z0334; RRID:AB_2314535), hAPP695 (6E10) (1:500; BioLegend #803001; RRID: AB_2564653), Iba1 (1:600; Wako #019–19741; RRID:AB_839504), AIF-1/Iba1 (1:600; Novus Biologicals #NB100-1028; RRID:AB_3148646), IL-1β (1:200; R&D #AF-401-NA; RRID:AB_416684), IL-18 (1:300; MBL #D047-3; RRID:AB_592016), IL-18R⍺/IL-1 r5 (1:100; R&D #AF856; RRID:AB_355664), MAP2 (1:500; Invitrogen #MA5-12,826; RRID:AB_10976831), NET (1:500; Atlas Antibodies #AMAb91116; RRID:AB_2665806), NLRP3 (1:200; Adipogen #AG-20B-0014; RRID:AB_2490202), p-NFκB (Ser536) (1:200; Cell Signaling #3033; RRID:AB_331284), S100β (1:500; SYSY #287 006; RRID:AB_2713986), SERT (1:500; Millipore #PC177L; RRID:AB_2122553), TH (1:1000; Millipore #MAB318; RRID:AB_2201528), TH (1:500; Millipore #AB152; RRID: AB_390204).
Secondary Antibodies (Thermo Fisher): Alexa Fluor-488 donkey anti-rabbit (1:200; #A-21206; RRID:AB_2535792), Alexa Fluor-555 donkey anti-rabbit (1:200; #A31572; RRID:AB_162543), Alexa Fluor-647 goat anti-rabbit (1:200; #A-31573; RRID:AB_2536183), Alexa Fluor-488 donkey anti-mouse (1:200; #R37114; RRID:AB_2556542), Alexa Fluor-555 donkey anti-mouse (1:200; #A31570; RRID:AB_2536180), Alexa Fluor-488 donkey anti-rat (1:200; #A21208; RRID:AB_2535794), Alexa Fluor-647 goat anti-rat (1:200; #A-21247; RRID:AB_141778), Alexa Fluor-488 donkey anti-goat (1:200; #A-11055; RRID:AB_2534102), Alexa Fluor-647 donkey anti-goat (1:200; #A-21447; RRID:AB_141844), Alexa Fluor-555 goat anti-chicken (1:200; # A-21437; RRID:AB_2535858).
Exclusively for the representative confocal images, after the quantitative analysis, LUTs were equally increased at the same level for all groups of a given experiment. Quantitative analyses were performed on raw images.
Immunofluorescence sections were used to estimate: TH+ neurons in the left SNpc, VTA and LC, 5-HT+ neurons in the entire IPN; 5-HT+ neurons in the dorsal (dRaphe) and medial Raphe (mRaphe); Iba1+ cells in the hippocampus, dorsal and ventral striatum and GFAP+ cells in the hippocampus. The area boundaries for VTA/SNpc/LC were defined by TH, the IPN/dRaphe/mRaphe by 5-HT, the dorsal and ventral striatum and hippocampus by DAPI staining, using the 5x-objective, in accordance to Paxinos guidelines [43].
We applied an optical fractionator stereological design using the Stereo Investigator System (MBF Bioscience). A stack of MAC5000 controller modules (Ludl Electronic Products, Ltd) was interfaced with a Zeiss Microscope Axio Imager KMAT with a motorized stage and a Zeiss Axiocam 506 mono with Working High End PC. A 3D-optical fractionator counting probe (x, y, z dimension 50 × 50 × 25 μm for TH+ neurons, 70 × 70 × 25 μm for 5-HT+ neurons and 100 × 100 × 25 μm for glia cells) was applied. Cells were marked with a 100x-oil-(VTA/SNpc) or a 40x-objective (neurons in LC/IPN/dRaphe/mRaphe; glia cells in hippocampus and striatum).
The total cell number was estimated according to the formula (Eq. 1):
$$text{N }=text{ SQ }times (1/text{ssf}) times (1/text{asf}) times (1/text{tsf})$$
(1)
where SQ represents the cell number counted in all optically sampled fields of the ROI, ssf is the section sampling fraction, asf is the area sampling fraction and tsf is the thickness sampling fraction.
Microglia were imaged with a Zeiss Microscope Axio Imager KMAT with motorized stage and a camera connected to Neurolucida software (7.5v; MBF Bioscience) for quantitative 3D-analysis of the entire cell [48, 49]. Only non-overlapping cells that showed clear soma and branching were analyzed. Soma area and perimeter were measured; Sholl analysis included counting the number of dendritic intersections, nodes and endings, and dendritic lengths at fixed distances from the soma in 10 μm-spaced concentric circles originating from the soma. Analysis was done with 100x-oil objective. Nine representative cells/animal were analyzed randomly and data were averaged for each mouse.
The ipsilateral hippocampus was dissected from the entire brain and stored at −80 °C until the day of the experiment. The ipsilateral midbrain was isolated from 500 μm-frozen slices using a biopsy punch. Tissue was homogenized in RIPA buffer containing (in mM): 50 Tris–HCl pH 7.5, 150 NaCl, 5 MgCl2, 1 EDTA, 1% Triton X-100, 0.25% sodium deoxycholate, 0.1% SDS, 1 sodium-orthovanadate, 5 b-glycerophosphate, 5 NaF and protease inhibitor cocktail; samples were then sonicated four times (five strokes of 0.5 pulse/s) and incubated on ice for 30 min [50]. Samples were centrifuged (13000 g, 20 min, + 4 °C) and the supernatant’s protein concentration was determined by the Bradford method. Proteins were applied to SDS-PAGE and electroblotted on a polyvinylidene-difluoride membrane. Blotting analysis was performed using a chemiluminescence detection kit. The relative levels of immunoreactivity were determined by densitometry using ImageJ.
Primary antibodies: Caspase-3 (1:500; Cell Signaling Technology; #9662s; RRID:AB_331439), Glycogen Synthase Kinase 3β (GSK3β; 1:1000; Cell Signaling Technology; #9832s; RRID:AB_10839406); p-GSK3β (Ser9) (1:1000; Cell Signaling Technology; #9336s; RRID:AB_331405); JNK (1:1000; Cell Signaling Technology; #9252s; RRID:AB_2250373); p-JNK (Thr183/Tyr185) (1:1000; Cell Signaling Technology; #9251s; RRID:AB_331659); p-p38 (Thr180/Tyr182) (1:1000; Cell Signaling Technology; #4511s; RRID:AB_2139682); p38 (1:1000; Cell Signaling Technology, #8690s; RRID:AB_10999090); TH (1:500; Abcam, #ab112; RRID:AB_297840); β-Tubulin (1:1000; Biolegend; #801201; RRID:AB_2313773). All primary antibodies were incubated overnight except for β-Tubulin (2 h).
Secondary antibodies: goat anti-mouse IgG (1:3000; Bio-Rad; #1706516; RRID:AB_11125547), goat anti-rabbit IgG (1:3000; Bio-Rad; #1706515; RRID:AB_2617112).
Membranes were stripped using Re-Blot Plus Strong Solution (Millipore; 15 min, RT). Both groups (Shan vs Casp3; Tg Sham vs Tg Casp3) were analyzed simultaneously.
Hippocampus from Sham and Casp3 CX3CR-1GFP mice (Sham: n = 3; Casp3: n = 5 mice) was dissociated into single-cell suspensions using Adult Brain Dissociation kit (Miltenyi Biotec, #130–107-677) according to the manufacturer’s instructions. Briefly, each mouse was transcardially perfused with 1 × D-PBS+/+. The hippocampus of each Sham or Casp3 CX3CR-1GFP mouse was isolated and added in half brain of a C57BL/6N mouse, to increase the quantity of tissues to dissociate. Tissues were then cut into small pieces and enzymatically and mechanically dissociated on the gentle MACS Octo Dissociator with Heaters (Miltenyi Biotec, #130–096-427) using the 37C_ABDK_01 program. Following dissociation, samples were resuspended and passed through a prewet 70 μm MACS SmartStrainer (Miltenyi Biotec, #130–110-916,) and centrifuged at 300 g for 10 min at 4 °C. Following centrifugation, the supernatant was discarded, and the debris were removed using the Debris Removal solution (Miltenyi Biotec, #130–109-398) provided by the kit. Finally, cells were resuspended in 100 μL of Running Buffer and stained for 30 min at 4 °C protected from light with the following antibodies: CD11b Monoclonal Antibody (M1/70) PE-Cyanine7 (1:100; Invitrogen, #25-0112-82; RRID: AB_469588) and PerCP anti-mouse CD45 Antibody (1:100; BioLegend #103130; RRID: AB_893339). To exclude dead cells, LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (1:100; Invitrogen, #L34957) was used. Cells were washed with Running Buffer and pelleted at 1000 g for 10 min at 4 °C. Then, cells were resuspended in 400 µL of Running Buffer. Afterward, the samples were taken to FACS and the gating strategy was set to CD11b+CD45+eGFP+ for isolation of microglia from Sham or Casp3 CX3CR-1GFP hippocampi. The sorting purity was to be approximately 97%.
Total RNA from isolated microglia was purified via the spin-colum based “Total RNA purification Plus Micro Kit” (Norgen Biotek, #48500), following the manufacturer instructions.
Low-input mRNA-Seq was performed by Igatech Service (Udine, Italy). Briefly, RNA samples were quantified, and quality tested by TapeStation RNA assay (Agilent Technologies). Libraries were generated using the Ovation SoLo RNA-seq Library Preparation kit (Tecan Genomics) following the manufacturer’s instructions. Final Libraries were checked with Qubit 3.0 Fluorometer (Invitrogen) and Agilent Bioanalyzer DNA assay. Libraries were then barcoded for multiplexing and sequenced on paired-end 150 bp mode on NovaSeq 6000 (Illumina) at a depth of 30 million reads. Base calling and demultiplexing was performed with Illumina BCL Convert v3.9.31. Lower quality bases and adapters were removed by Cutadapt v1.11 software [51]. Reads were aligned on reference GRCm39.112 genome with STAR [52] using default parameters. Assembling and quantitation of full-length transcripts representing multiple spliced variants for each gene locus was performed by Stringtie [53] using default parameters.
Differentially expressed genes (DEGS) were identified with DESeq2 [54]. The ToppGene suite was used with default settings to identify functional enrichment in the DEG lists (http://toppgene.cchmc.org).
Quantification of hippocampal monoamines (DA, 5-HT, NE), and relative metabolites (DOPAC and HVA for DA, 5-HIAA for 5-HT, MOPEG for NE) was performed using an HPLC system (UltiMate® 3000, Thermo Fisher) coupled with Coulochem electrochemical detection (6011RS Ultra Coulometric Analytical Cell, Thermo Fisher). After animal sacrifice, the left hippocampus was rapidly dissected on ice and immediately stored at −80 °C until analysis. On the day of analysis frozen samples were homogenized on ice with 0.05 M HClO4 and antioxidant solution (containing in mM: 0.27 Na2EDTA, 100 acetic acid, 0.0125 ascorbic acid) in a 4:1 ratio. The homogenate was mechanically lysed, sonicated on ice and centrifuged (10000 rpm, 20 min, 4 °C). The supernatant was transferred into a new tube and the pellet was weighed. 20 µL of each sample were injected onto the HPLC-ECD (run-time: 60 min; flow-rate: 0.6 mL/min). Standards of each metabolite were prepared fresh on the day with same solutions and quantities as for tissue samples. The chromatographic separation was performed on a Hypersil GOLD aQ-C18 column (150 × 3 mm, 5 µm) fitted with an aQ-C18 drop-in guard pre-column (10 × 3 mm, 5 µm) maintained at 37 °C. The mobile phase consisted of 5% methanol and buffer solution (0.1 M Na-Phosphate, 0.1 mM Titriplex® III and 0.5 μM 1-Octanesulfonic-Acid Na-salt, pH 3.6 adjusted with 85% Ortho-Phosphoric-acid), filtered through a 0.22 µm cellulose-ester membrane (Millipore). The potential applied to both dual-inline flow-through micro-porous graphitic carbon-working electrodes was set at + 450 mV, with 1 nA-gain for ECRS1 and 10 nA-gain for ECRS2. The chromatograms were integrated with Chromeleon™ Software (7.0v; Thermo Fisher). The sample concentration of metabolites was calculated from the corresponding peak height, normalized to the pellet weight.
Following head dislocation, mice were decapitated and the brain was rapidly removed; 300 μm-thick parasagittal slices containing the left dorsal hippocampus were cut (Leica VT1200S vibratome) in oxygenated (95% O2, 5% CO2) ice-cold sucrose-based solution (in mM: 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 MgSO4, 0.5 CaCl2, 25 glucose, 185 sucrose; ~ 300 mOsm, pH 7.4). Brain slices were left to recover in artificial Cerebro-Spinal Fluid (aCSF; containing in mM: 124 NaCl, 1.25 NaH2PO4-H2O, 26 NaHCO3, 3 KCl, 10 Glucose, 1 MgSO4, 2 CaCl2, ~ 300 mOsm, pH 7.4) at 32–34 °C for 40 min, and moved at RT for at least 30 min before recording [55].
For recording, each slice was placed under an Olympus BX51WI microscope and perfused with oxygenated aCSF (3-4 mL/min, 30-32 °C). The hippocampus was visualized with infrared differential-interference-contrast at 4x.
Recordings were performed using a MultiClamp700B amplifier, digitized with Digidata1550B and computer-saved with pClamp11 (Molecular Devices). Patch-pipettes (3–5 MΩ), filled with aCSF, were pulled from TW150F-4 glass tubes (WPI). Field excitatory postsynaptic potentials (fEPSPs), recorded in the stratum radiatum, were induced with Schaffer collateral stimulation (100 μs), acquired at 20 kHz and filtered off-line with a 10 kHz low-pass Bessel filter. Input–Output (I/O) curves of fEPSP slopes were obtained at 10 μA-stepped increasing stimulation every 30 s. The Paired-Pulse Ratio (PPR) was evaluated with pairs of stimuli (20–1000 ms interval), at half-maximal stimulation. For Long-Term Potentiation (LTP), after 20 min of test stimulation (half-maximal intensity, every 30 s) to assess slope stability, the slice was challenged with two trains at 100 Hz (1 s duration, 20 s interval) followed by test stimulation for 1 h. The LTP magnitude was evaluated as the fEPSP mean slope at 60 min after the conditioning trains, normalized to the mean baseline slope [45, 56, 57]. To assess the effect of the IL-1β receptor antagonist (IL-1β-Ra) on LTP, slices were pre-incubated with IL-1β-Ra (100 ng/mL, from freshly-thawed stock in PBS/bovine-serum albumin 0.1%; R&D Systems) in oxygenated aCSF for 1 h and recorded as above.
Coronal slices (240 μm-thickness) were used for spine density analysis. CA1 pyramidal neurons were filled with biocytin during whole-cell patch clamp. Freshly weighted biocytin (0.2%; Tocris) was added to intracellular solution containing (in mM): Cs-methanesulfonate 120, CsCl 15, NaCl 8, HEPES 10, EGTA 0.20, TEA-Cl 10, QX314-Cl 5, Mg-ATP 2, Na-GTP 0.3 (~ 275–285 mOsm, pH 7.4). Patch-pipettes used for cell filling had a tip resistance of 4-10 MΩ and a minimum filling time of 10 min was used for each cell. Access resistance was monitored throughout this time to check for accessibility to the cytoplasm. To avoid cell damage after filling, the electrode was removed from the slice by establishment of an outside-out patch.
For analysis of biocytin-filled neurons, slices bearing neurons loaded with biocytin were fixed by immersion in 4% paraformaldehyde in PB (overnight, 4 °C). Then, slices were washed three times in PB and incubated with Streptavidin (1:750; Alexa Fluor 555-conjugated #S32355, Thermo Fisher; RRID:AB_2571525) and Green-Fluorescent Nissl-Stain (NeuroTrace 1:600; Invitrogen #N21480) in PB containing 0.3% Triton X-100 (overnight, 4 °C).
The Z-stack confocal images were captured using Nikon Eclipse Ti2 confocal microscope with 60x-oil objective and 4.5 digital zoom. We analyzed spines in the distal (terminal) apical dendrites.
Spine density was assessed by counting the number of spines in at least 18 segments per neuron (4 cells, 5 animals per experimental group) chosen in branch orders 2–4 of apical dendrites extending for an additional 30–60 μm away from the starting point [58]. Imaris software (9.8.2) was utilized to create 3D reconstructions of dendritic segments using the Filaments tool. The settings used for all reconstructions is the same as in [59]. An observer blinded to the experimental groups manually edited the reconstructions to include or exclude misidentified spines.
All behavioral tests were conducted between 09:00 a.m. and 04:00 p.m.. Mice were habituated to the experimenter through daily handling sessions for one week prior to the onset of behavioral testing. In addition, animals were acclimated to the testing room for one hour immediately before the start of each experimental session.
To minimize potential olfactory interference, the chambers and objects were cleaned between animals and/or sessions using a 5% ethanol solution.
Testing was performed in a dimly-lit (25 lx) plexiglass open field arena (60 × 60 × 30 cm), with dark-grey walls and white floor. On Day1 (D1), each mouse was placed in the arena center and allowed to freely-explore for 10 min, during which movements were recorded. We analyzed the time spent (s) in the arena center and periphery, and the total distance traveled. Thereafter, we conducted the NOR test, consisting of habituation, training and testing [60]. During habituation, mice were familiarized with the empty arena for 10 min (D2). 24 h later (D3), mice were trained for 10 min by exposure to two identical objects (yellow wooden spheres) placed in the arena center. Mice were then returned to their home cage. Following 24 h (D4), mice were returned to the arena for the test session (10 min), during which one object was replaced by a novel object (a light-grey wooden cone). In both the training and testing sessions the animals were left to freely-explore the objects, and the exploration time was recorded, calculated as the time when they touched or climbed on an object or sniffed it at a distance of at least 2 cm. The Object Discrimination Ratio (ODR) was calculated using the following formula (Eq. 2):
$$ODR= frac{text{Time exploring the novel object}}{text{Time exploring novel object }+text{ Time exploring familiar object}}text{ x }100$$
(2)
Objects were randomized and counterbalanced across groups.
The SOR test was conducted in the same circular arena previously described for the open field and NOR tasks. For this test, a high-contrast black-and-yellow striped cue was affixed to the inner wall of the arena to provide a visual local cue and enhance spatial orientation. Each mouse was tested individually across five consecutive 6-min sessions, with each session separated by a 3-min inter-trial interval in the home cage. In all sessions, animals were placed into the arena from the same starting location (protocol adapted from [61, 62]).
During session 1 (S1), mice were allowed to freely explore the empty apparatus, enabling familiarization with the environmental context and distal cues in the absence of objects. During sessions 2 to 4 (S2-S4), mice were exposed to four distinct objects (A-D) placed in fixed positions to allow the animals to encode the spatial configuration (habituation phase). The objects differed in shape, size, and material to promote discrimination and reduce object bias: (A) a yellow wooden ball, (B) a gray metal column, (C) a light-grey wooden cone, and (D) a glass jar with a perforated red cap.
Habituation to the spatial configuration was assessed by averaging the duration of contact with the four objects during sessions 2, 3, and 4 for each experimental group to provide an index of general exploratory activity. Object exploration was defined as the total time (s) the animal spent with its snout either in direct contact with the object or within approximately 2 cm of it.
Session 5 (S5) involved a spatial rearrangement: object A was moved to the previous location of object B, object B was placed in a novel, unoccupied position, while objects C and D remained in their original locations. This configuration was used to assess spatial novelty detection based on differential exploration of Displaced Objects (DO) versus Non-Displaced Objects (NDO), using the following formula (Eqs. 3 and 4):
$$text{DO }left[text{S}5right]-text{DO }left[text{S}4right]=text{DO}$$
(3)
$$text{NDO }left[text{S}5right]-text{NDO }left[text{S}4right]=text{NDO}$$
(4)
These values were used to quantify the change in exploratory behavior elicited by the spatial rearrangement.
All sessions were video-recorded using a ceiling-mounted camera, and behavioral tracking was performed offline with EthoVision XT v.17.0 (Noldus).
Motor coordination and motor learning were assessed using the accelerated rotarod test. The experimental procedure, performed over three consecutive days, consisted of two days of training followed by one test session on the third day (protocol adapted from [63]). The test was conducted using a computerized four-lane rotarod apparatus (Panlab, Harvard Apparatus), featuring a rotating cylinder (3 cm diameter) with independently controlled lanes, such that up to four mice could be tested simultaneously.
On the first day, mice underwent an acclimation phase consisting of a single 30 s trial on the rod rotating at a constant speed of 4 rpm. Subsequently, in each session, the rod acceleration was programmed to increase Linearly from 4 to 40 rpm over a 300 s period. Each session included four trials per mouse, separated by 5 min inter-trial intervals. The following behavioral parameters were measured for each trial: latency to fall (time in seconds spent on the rod) and the best performance across sessions, defined as the longest latency to fall on the final test day.
The sample number per group and experiment was determined by power analysis (G*Power software, 3.1.9.7v) using a power of 0.8 and errors of 0.05; standard deviations of all groups were obtained from previous publications with similar experiments.
Randomization was done with a random-number table to decide how mice from the same litter would be randomly assigned to the different groups.
Researchers were blinded to the animal group; un-blinding occurred after analysis.
The experimental units for each experiment are described in detail in Figure legends.
Analyses were performed using Prism8.01 (GraphPad). Data were checked for normality using the Shapiro–Wilk and D’Agostino-Pearson tests. Data from two groups (i.e., Sham/Casp3, Saline/6OHDA, WT/Tg2576, Tg Sham/Tg Casp3) were analyzed with Two-tailed parametric (unpaired t-test or Welch’s t-test) or non-parametric Mann–Whitney tests according to normality.
Sholl analysis data, PPR, I/O curves, mean exploration time during the training and test phases of the NOR test, mean exploration time during the familiarization and test phases of the SOR test, and mean latency to fall across sessions in the rotarod test were analyzed by Two-way Repeated-Measures (RM) ANOVA, using distance from soma, interval, stimulus intensity, object category (left vs right and novel vs familiar), test sessions (S1-4), object category (DO vs NDO), and number of sessions (S1-12) as repeated values, respectively. Post-hoc tests were performed using Sidak’s or Tukey’s multiple comparison tests.
Data from three or four groups were analyzed by One-Way ANOVA followed by Tukey’s multiple comparison test, or with Kruskal–Wallis followed by Dunn’s multiple comparison test.
For the differential expression analysis of transcriptomics expression, statistical significance was determined using the Wald test [64]. Cut offs (p < 0.050) were used for p values and for p values adjusted with the False Discovery Rate (FDR) methods of Benjamini–Hochberg and Benjamini-Yekutieli (FDR B&H and FDR B&Y, respectively).
See Figure legends for more details. p ≤ 0.05 indicates statistical significance. In box-and-whisker plots, the central line denotes the median, edges are upper and lower quartiles, whiskers show minimum and maximum values and points are individual experiments. In the violin plots used for spine density, the width of the plot at any given point corresponds to the frequency of data points at that value. The volcano plot for the transcriptomic analysis shows scattered individual data points. All other data are presented as mean ± s.e.m..

By Stuart Condie
SYDNEY--Australia's ANZ Group scrapped its share buyback and plans to invest in mortgage and commercial bankers as its new CEO tries to boost productivity and returns.
ANZ on Monday said it would halt its ongoing share buyback, which has about 800 million Australian dollars, or US$518 million, remaining. This will allow it to return about A$1 billion in surplus capital to the bank, it said.
ANZ, which is Australia's fourth-largest bank by market capitalization, will invest heavily in its in-house mortgage sales force with the aim of increasing the number of lenders in its branches by up to 50%.
It also wants to increase the number of commercial bankers servicing its business and private customers by up to 50%. It plans to develop its own pipeline of bankers in a new Commercial Bankers Academy.
"Our people will deliver our strategy, and we must focus on a culture of customers, performance and talent," said Chief Executive Nuno Matos, who took charge this year.
Write to Stuart Condie at stuart.condie@wsj.com
(END) Dow Jones Newswires
October 12, 2025 19:08 ET (23:08 GMT)
Copyright (c) 2025 Dow Jones & Company, Inc.

A new University of Michigan study offers one of the most detailed pictures so far of how violent sun storms spread across space — and occasionally, straight to our planet. With the help of supercomputer simulations, researchers traced the…

If you want to take a real-time sample of the global economy in action at any one moment, then the Cathay Cargo Hub at Hong Kong International Airport (HKG) is a particularly fascinating node.
HKG is the world’s busiest air-cargo hub and no airline moves more freight through it – more than 1.7 million tonnes annually – than Cathay Pacific. Founded in 1946 as a cargo operator between Australia and China, Cathay now manages a sprawling 246,000 sq m hub and the constant hum of palletised and shrink-wrapped goods. In just one section of the warehouse, the inventory ranges from huge cases of Château d’Arche wine to Golden Lily mangoes from the Philippines, alongside priceless artworks packed in Cadogan Tate crates – all destined for the holds of windowless cargo freighters.
Air cargo accounts for less than one per cent of all goods shipped by tonnage globally but it makes up some 35 per cent of the total value. It carries all the things the world needs or wants in a hurry, from the latest iPhone and cold-chain pharma to Italian supercars and oil-drilling machinery. This urgency is just one of the reasons why air cargo is worth paying attention to: when people stop buying the things they need, it’s often a reliable signal that a recession might be on the horizon.
On a recent weekday morning, the warehouse was particularly active, catching up from the two-day stoppage enforced by Super Typhoon Ragasa, a level-10 storm that sideswiped Hong Kong. But the air-cargo world faces headwinds beyond the weather, including the unpredictable currents of geopolitics.
Not long ago, air cargo prices were being driven upwards as a result of “fast fashion” purchases and other inexpensive goods from Chinese companies such as Shein and Temu. But after the Trump administration eliminated de minimis exemptions on goods worth less than $800 (€688) – precisely what Shein and Temu had exploited – US-bound air cargo from China – a key, typically one-way route – fell by at least 25 per cent.
“Supply chains don’t change overnight,” says Tom Owen, cargo director at Cathay Cargo. “But there has been a recalibration.” One benefit for air-cargo companies such as Cathay is that they don’t own factories – they own planes. This agility can work in their favour, as Cathay Group CEO Ronald Lam noted at the Routes World conference in Hong Kong, the day after the typhoon blew through. The uncertainty of tariffs, he said, created a “rush” to place orders. Air cargo, with its far shorter lead times compared with ocean shipping, “is really well positioned to capture that rush.” Hong Kong’s position as a global hub means that Cathay Group can adjust its routes swiftly. “We are getting more cargo from India and Southeast Asia, routed via Hong Kong to the US, to replenish the slowdown on US-bound cargo from China.”
During a week when the Trump administration threatened yet again to escalate the trade war that it began, air-cargo companies such as Cathay find themselves in the eye of the storm. But this is an industry in which agility and flexibility are key. During the coronavirus pandemic, cargo kept many airlines afloat as they quickly converted their passenger fleet into impromptu freighters, loading cargo where passengers normally sat. The latest disruptions present a new challenge but also a lesson. While long-term planning is crucial to success in business, the best strategies leave room to adapt. Take it from Cathay, which has dealt with its fair share of turbulence: be ready to change course quickly, even while mid-air.
Tom Vanderbilt is a regular Monocle contributor. For more opinion, analysis and insight, subscribe to Monocle today.


At the age of 15, medics feared Katie Mitchell was coming to the end of her life after suffering irreversible lung damage and heart failure from a rare congenital disease.
But she defied the odds thanks to a heart and lung transplant, and at the…

“The Inbetweeners,” the hit British teen comedy series that gave the world a foul-mouthed library of hilariously puerile quotes not to mention two hugely successful spin-off movies, looks set to make a comeback.
Banijay UK and Fudge…
This request seems a bit unusual, so we need to confirm that you’re human. Please press and hold the button until it turns completely green. Thank you for your cooperation!